News
No Fear of Superiority, Non-Inferiority, and Equivalence Studies!
Understanding the science of data
This article was published in MedinLux and is part of a collaborative effort to make statistical and epidemiological concepts accessible to healthcare professionals in Luxembourg.
In one of our previous articles, we described the main principles of clinical trial design. Here, we focus on three types of designs mainly used in phase III clinical trials: superiority, non-inferiority, and equivalence studies.
Phase III trials are comparative studies, generally randomised, involving several hundred or even thousands of participants. Their objective is to evaluate the efficacy and safety of a new treatment or a medical device on a large scale, in comparison with a reference treatment or a placebo. When the results are considered favorable, they form the basis of the Marketing Authorisation Application submitted to regulatory authorities.
The choice of study design determines the entire protocol: statistical hypotheses, sample size calculation, analysis methods, and interpretation of the results.
“Outcomes” and “Endpoints”: Definitions and Challenges
First and foremost, defining the evaluation criteria precisely is essential. These elements are described in the protocol and outlined in the statistical analysis plan. Although the terms “outcome” and “endpoint” are often used interchangeably, they refer to distinct concepts:
- “Outcome”: any measure related to participants’ health status (depression score, tumor size, blood pressure, occurrence of a cardiovascular event, etc.).
- “Endpoint”: the time-specific and predefined “outcome” measure used to address the primary objective of the study (for example: reduction in systolic blood pressure between baseline and week twelve).
A relevant “endpoint” must have clear clinical relevance and be defined with sufficient precision to ensure reproducibility of the measurement. It must be consistent with the sample size calculations and, whenever available, rely on validated measuring tools. The a priori definition of the primary endpoint is essential to avoid any post hoc decisions on interpretation of the results, as those may bias their interpretation. The nature of the “endpoint” (continuous, binary, time-to-event) will also influence the choice of statistical methods and study design.
Superiority Study
The superiority study is the most classical design in clinical research. Its objective is to demonstrate that the new treatment is statistically superior to the comparator, whether this comparator is a placebo or another active treatment.
In this context, the statistical hypotheses assessed in the study are defined as follows:
- Null hypothesis: there is no statistically significant difference, measured by the “endpoint,” between the new treatment and the control treatment at the alpha risk level (see our article on the p-value or the box);
- Alternative hypothesis: the difference between the two treatments is statistically significant.
The analysis is typically performed using two-sided 95% confidence intervals (95% CI). Superiority is demonstrated when this confidence interval does not include the value indicating the absence of effect (0 for an absolute difference, 1 for a ratio such as a relative risk, odds ratio, or hazard ratio). Some fictional examples:
- a mean difference in systolic blood pressure of 6 mmHg (95% CI: -10 mmHg – -2 mmHg) following treatment administration → the CI does not include 0 → statistically significant difference at the 5% level;
- a survival comparison after a cancer diagnosis, comparing a new treatment with the standard treatment, yielding a hazard ratio (HR) of 0.78 (95% CI: 0.65–0.92) → the CI does not include 1 → significant reduction in risk with the treatment;
- occurrence of stroke, comparing two populations receiving either a new treatment or the standard treatment, with an HR of 0.85 (95% CI: 0.65–1.10) → the CI includes 1 → no demonstrated superiority of the new treatment.
The primary analysis is generally conducted according to the intention-to-treat principle (ITT), meaning that participants are analysed according to the group to which they were initially randomized, regardless of the treatment actually received. This preserves the benefits of randomization and provides a more pragmatic estimate of the treatment effect.
When interim analyses are planned, they must be specified in advance and utilise methods for adjusting the alpha risk in order to limit inflation of the type I error risk (see our article on the p-value). An interim analysis may be necessary to evaluate major efficacy, obvious lack of efficacy (futility), safety concerns, or a revision of the sample size. In such cases, continuation of the trial may raise ethical issues and lead to early termination of the study.
- The α risk (alpha) corresponds to the statistical significance threshold, usually set at 0.05 in clinical trials. This means that researchers accept a maximum 5% risk of incorrectly concluding that a difference between the treatment and placebo groups exists (type I error). Saying that there is no significant difference at the α level means that the p-value is above the chosen threshold (often p > 0.05).
- Caution: this does not necessarily mean that the two treatments are equivalent, but only that the study did not demonstrate a difference. This may have occurred due to insufficient statistical power, a sample size that is too small, or an existing but too small effect to be detected.
Non-Inferiority Study
The non-inferiority design addresses a different question. The aim is no longer to demonstrate that the new treatment is better, but instead that it is not clinically inferior to the reference treatment (beyond a clinically acceptable loss of efficacy). This type of study is particularly relevant when the use of a placebo is not ethically acceptable, especially when an effective standard treatment already exists. It is also chosen when the new treatment offers other advantages, such as better tolerability, easier administration, or lower cost. The choice of this design is therefore driven by the balance between efficacy and other patient-relevant benefits.
The definition of the non-inferiority margin, denoted M, is the central element of this design. This margin must be pre-specified before the start of the study, be based on historical evidence regarding the efficacy of the reference treatment versus placebo, and be statistically valid. It must represent a clinically relevant proportion of the previously demonstrated effect and should not neither too small nor too large. Its determination requires close collaboration between clinicians and biostatisticians (FDA document: Non-Inferiority Clinical Trials to Establish Effectiveness – Guidance for Industry).
From a statistical perspective, the hypotheses tested in the study are:
- Null hypothesis: the difference between the control treatment and the test treatment is greater than or equal to the non-inferiority margin M;
- Alternative hypothesis: the difference is strictly less than M.
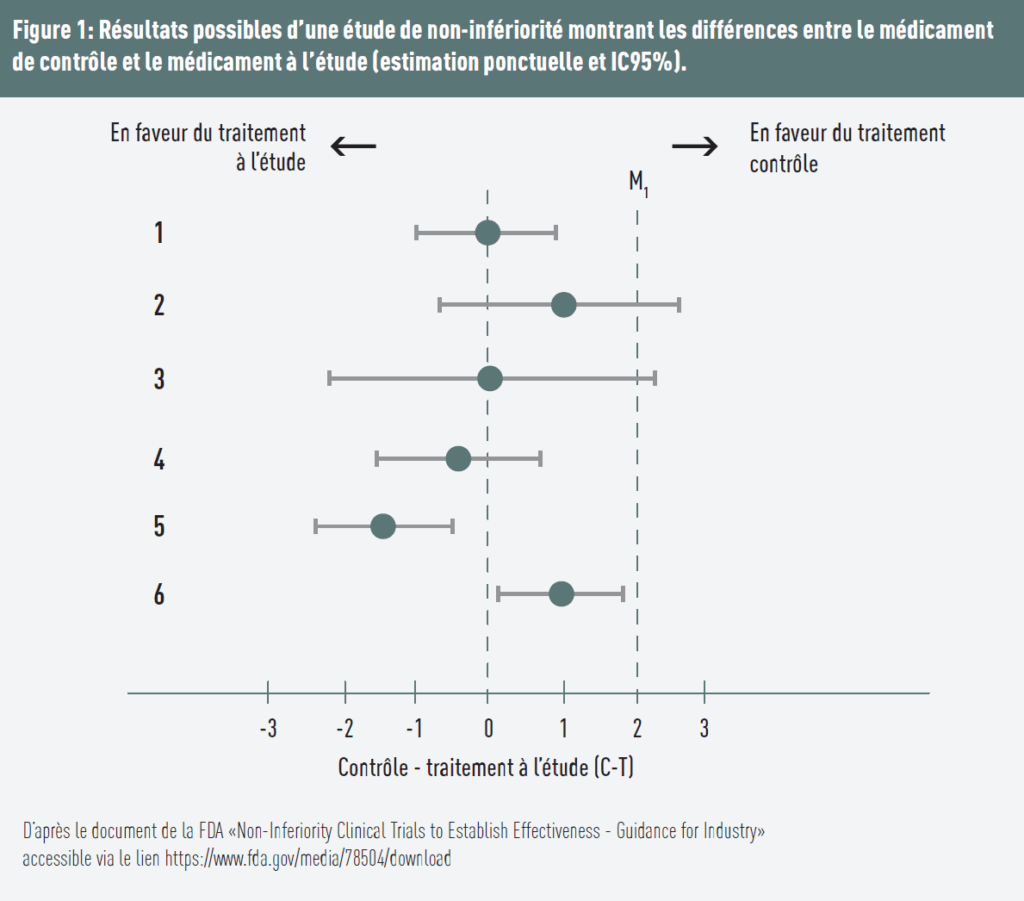
Several scenarios are illustrated in Figure 1:
- The difference equals 0, assuming identical effects of both treatments. The upper bound of the 95% confidence interval is below the predefined threshold → non-inferiority is demonstrated;
- The control treatment is more favorable, and the upper bound of the confidence interval is above the threshold → non-inferiority is not demonstrated;
- The difference is 0, but the upper bound of the confidence interval is above the margin → non-inferiority is not demonstrated;
- The test treatment is favored (estimate < 0) and the upper bound of the confidence interval is below the margin → non-inferiority is demonstrated;
- The test treatment is favored, and the upper bound is below both the margin and 0 → both non-inferiority and superiority are demonstrated;
- The control treatment appears more effective (positive estimate), but the entire confidence interval remains below the margin Δ → non-inferiority is demonstrated. However, since the lower bound of the confidence interval is above 0, the results also suggest that the test treatment is statistically inferior to the control treatment. This means that the test treatment is significantly worse but not intolerably worse.
Recommendations from the European Medicines Agency (EMA) and the US Food and Drug Administration (FDA) emphasize the importance of conducting the intention-to-treat (ITT) analysis. In superiority trials, ITT tends to dilute differences between groups and is therefore conservative. In non-inferiority trials, however, this dilution may artificially favour a conclusion of non-inferiority. For this reason, methodological guidelines generally recommend performing both per-protocol (PP) and ITT analyses.
Sample size calculation in this type of study strongly depends on the chosen margin. The smaller the margin, the larger the number of participants required to ensure sufficient statistical power. Sample size is also influenced by other factors such as the effect size and the number of participating centres. But that is another story.
In other words, M corresponds to the maximum acceptable loss of efficacy of the new treatment compared with the reference treatment, while still preserving a clinically relevant fraction of the effect of the latter. It therefore represents the statistical boundary below which the new treatment would be considered clinically unacceptable. As long as the estimated effect does not cross this limit, non-inferiority can be concluded.
PRACTICAL APPROACH:
- the first step is a literature review of the efficacy data for the reference treatment, usually derived from placebo-controlled trials;
- effect estimates and their confidence intervals are then combined;
- the margin is defined either based on the pooled effect or on the confidence interval bound closest to the null effect.
Other methods exist, but they are more complex.
Equivalence Study
The equivalence study aims to demonstrate that the effect of the new treatment is sufficiently close to that of the reference treatment to be considered clinically equivalent. Statistically, this translates into a difference between the two treatments lying within a predefined interval from –M to +M (see the definition of margin M described in the non-inferiority study). The objective is therefore to establish that the two treatments do not differ statistically in any direction.
In this framework, the null hypothesis corresponds to observing a difference lying outside the equivalence interval, while the alternative hypothesis corresponds to a difference lying within this interval. Equivalence is demonstrated when the entire 95% confidence interval lies within the pre-specified bounds. The test is conducted in a two-sided manner, which distinguishes it from non-inferiority analysis.
This design is frequently used when evaluating new formulations or changes in route of administration of the same drug. It should be emphasized that a superiority trial failing to show a statistically significant difference does not in any way allow one to conclude equivalence. The absence of evidence of a difference is not evidence of absence of a difference, particularly when the study was not powered to test this specific hypothesis.
The correct interpretation of various comparative trials requires close collaboration between clinicians and biostatisticians from the very beginning, from the design stage of the study.
OTHER CHARACTERISTICS OF STUDY DESIGNS EXIST
Clinical trial design types are not limited to the dimension previously presented in this article. In certain situations, other design parameters can be altered to better address the research question. For example, in a “cross-over” trial, each patient receives both the treatment and the placebo sequentially: they therefore act as their own control, which reduces inter-individual variability. In a factorial trial, multiple interventions are evaluated simultaneously, either alone or in combination, in order to study both their individual effects and potential interactions.
The development of a new drug or a vaccine remains a long process, typically lasting 15 to 20 years under standard conditions. To accelerate this process, more flexible designs have been developed. Some involve combining or covering multiple trial phases (for example phase I–II or II–III), in order to save time without compromising the assessment of safety and efficacy. This approach was widely used during the development of mRNA vaccines against COVID-19, where early phases were overlapped and phase III was initiated very rapidly based on interim data. Other so-called adaptive designs allow certain elements of the study to be modified during the trial based on interim analyses (for example adjusting doses, reallocating patients, or early stopping of an ineffective arm).
The COVID-19 pandemic also contributed to the popularisation of platform trials. These trials rely on a common infrastructure in which multiple treatments can be evaluated in parallel, with the possibility of adding or removing treatments over time. This design substantially improves the efficiency and responsiveness of clinical research.
Finally, the development of personalised medicine has led to the emergence of innovative designs. “Basket” trials evaluate a single targeted treatment across different diseases sharing a common molecular alteration. In contrast, “umbrella” trials test multiple targeted treatments within a single disease, based on the molecular characteristics of patient subgroups. These approaches allow for more precise adaptation of treatments to patients’ biological profiles.
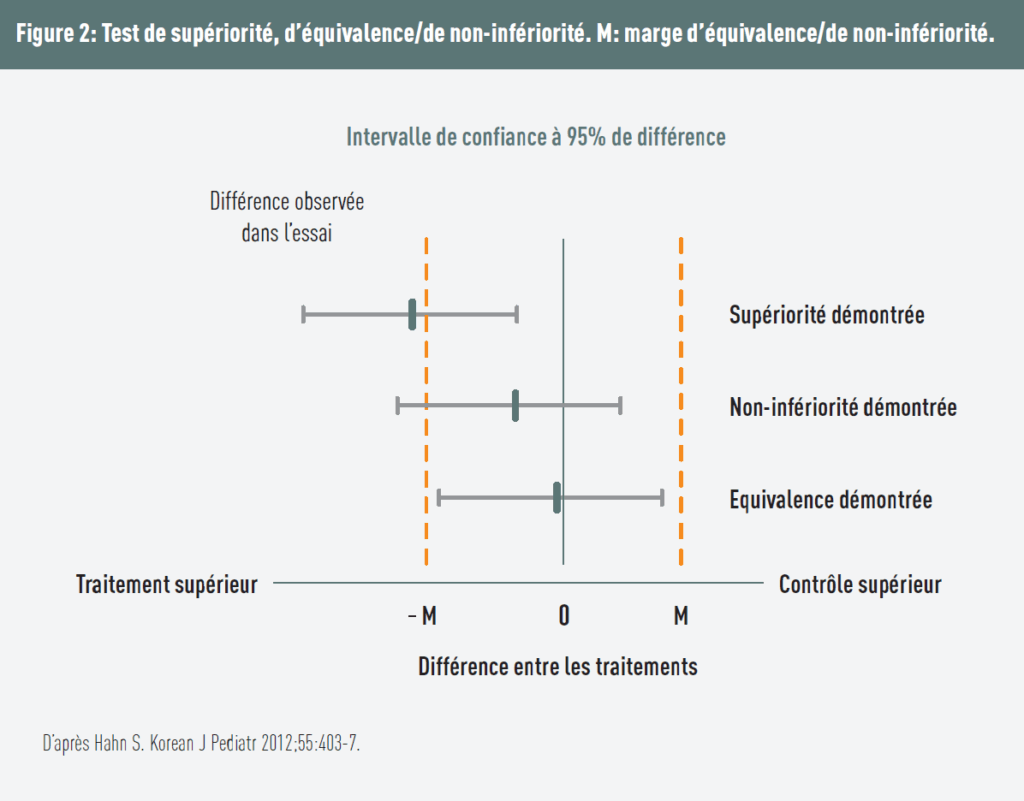
Conclusions
Figure 2 makes it possible to distinguish between the different study designs.
The margin M must not be confused with statistical significance: these are two completely different concepts. The margin is defined based on literature data, clinical experience, and discussions with clinicians; it is difficult to determine. The conclusions of non-inferiority and equivalence trials are based exclusively on this margin. A margin that is too wide may lead to accepting a clinically unacceptable loss of efficacy, whereas a margin that is too strict makes statistical significance particularly difficult to demonstrate.
The choice of the alpha risk determines the bounds of the confidence interval (an alpha risk of 5% corresponds to a 95% confidence interval). In a non-inferiority study, an alpha of 1% (and a power of 90% instead of 80%) may be chosen when a more conservative approach is sought (99% confidence interval). Non-inferiority then becomes more difficult to demonstrate, but the risk of a false positive is reduced, i.e., the chance that a new treatment is incorrectly considered non-inferior to the reference treatment is lower.
The choice of design must be made before the start of the study, as it determines the formulation of hypotheses, sample size calculation, analysis methods, and interpretation of results. The central question remains primarily clinical:
- Is my new treatment better than the existing treatment? → superiority trial
- Is my new treatment almost as effective, while offering other advantages? → non-inferiority trial
- Is my new treatment clinically equivalent to the existing treatment? → equivalence trial
Given the methodological and regulatory complexity of these comparative trials, early involvement of a biostatistician is essential to ensure the scientific validity and robustness of the conclusions.
Take-home messages
- The choice of trial design is primarily a clinical decision. It depends on the question being asked: demonstrating superiority, preserving existing efficacy, or showing equivalence.
- The absence of a statistically significant difference does not mean that two treatments are equivalent. A negative superiority trial does not allow conclusions about equivalence or non-inferiority.
- In non-inferiority and equivalence trials, the definition of the Δ margin is a critical step. A margin that is too wide may lead to accepting a clinically unacceptable loss of efficacy.
- The conclusions of non-inferiority and equivalence trials are essentially based on the confidence interval and the non-inferiority/equivalence margin.
- In non-inferiority trials, per-protocol and intention-to-treat analyses results should generally agree to strengthen the robustness of the conclusions.
- The correct interpretation of comparative trials requires close collaboration between clinicians and biostatisticians from the very design stage of the study.
The epidemiostatistical series published in MEDINLUX.
Related News