Actualités
No Fear of Superiority, Non-Inferiority, and Equivalence Studies!
Comprendre la science des données
Cet article a été publié dans MedinLux et fait partie d’un effort de collaboration visant à rendre les concepts statistiques et épidémiologiques accessibles aux professionnels de la santé au Luxembourg.
Lors d’un de nos précédents articles, nous avons décrit les grands principes du design des essais cliniques. Nous nous intéressons ici à trois types de schémas utilisés principalement dans les essais cliniques de phase III : les études de supériorité, de non-infériorité et d’équivalence. Les essais de phase III sont des essais comparatifs, généralement randomisés, incluant plusieurs centaines voire des milliers de participants. Leur objectif est d’évaluer l’efficacité et la sécurité d’un nouveau traitement ou dispositif médical à grande échelle, en comparaison avec un traitement de référence ou un placebo. Lorsque les résultats sont jugés favorables, ils constituent la base du dossier de demande d’Autorisation de Mise sur le Marché (AMM) auprès des autorités réglementaires. Le choix du schéma d’étude conditionne l’ensemble du protocole : hypothèses statistiques, calcul de la taille d’échantillon, modalités d’analyse et interprétation des résultats.
« Outcomes » et « endpoints » : définitions et enjeux
Avant toute chose, la définition précise des critères d’évaluation est essentielle. Ces éléments sont décrits dans le protocole et détaillés dans le plan d’analyse statistique. Bien que souvent utilisés de manière interchangeable, les termes « outcome » et « endpoint » renvoient à des concepts distincts :
- « Outcome » : toute mesure liée à l’état de santé d’un participant (score de dépression, taille de la tumeur, pression artérielle, survenue d’un événement cardiovasculaire, …).
- « Endpoint » (critère d’évaluation) : définition opérationnelle et temporelle de l’« outcome » servant à répondre à l’objectif principal de l’étude (par exemple : diminution de la pression artérielle systolique entre le début de l’étude et la douzième semaine).
Un « endpoint » pertinent doit présenter un intérêt clinique évident et être défini de manière suffisamment précise pour garantir la reproductibilité de la mesure. Il doit être cohérent avec le calcul de la taille d’échantillon et, lorsque cela est possible, reposer sur des instruments validés. La définition a priori du critère principal d’évaluation est essentielle afin d’éviter toute interprétation a posteriori des résultats, ce qui fausserait l’interprétation. La nature de l’« endpoint » (continu, binaire, temps jusqu’à l’événement) influencera également le choix des méthodes statistiques et du design.
Etude de supériorité
L’étude de supériorité constitue le design le plus classique en recherche clinique. Son objectif est de démontrer que le nouveau traitement est statistiquement supérieur au comparateur, qu’il s’agisse d’un placebo ou d’un traitement actif.
Dans ce cadre, les hypothèses statistiques évaluées dans l’étude sont définies comme suit :
- Nulle : il n’existe pas de différence statistiquement significative, mesurée par l’«endpoint», entre le nouveau traitement et le traitement de contrôle au risque alpha (voir notre article sur la p-valeur et l’encadré);
- Alternative : la différence entre les deux traitements est statistiquement significative.
L’analyse se fait de façon bilatérale avec des intervalles de confiance à 95% (IC95%). La supériorité est démontrée lorsque cet intervalle de confiance n’inclut pas la valeur correspondant à l’absence d’effet (0 pour une différence absolue, 1 pour un ratio tel qu’un risque relatif, un odds ratio, un hazard ratio). Quelques exemples fictifs:
- une différence moyenne de la pression artérielle systolique égale à 6mmHg (IC95%:-10;-2), suite à la prise d’un traitement → l’IC n’inclut pas 0 → différence statistiquement significative au risque de 5%;
- une comparaison de survie suite à un diagnostic d’un cancer, comparant un nouveau traitement avec le traitement standard, obtenant un hazard ratio (HR) de 0,78 (IC95%: 0,65–0,92) → l’IC n’inclut pas 1 → réduction significative du risque avec le traitement;
- la survenue d’accidents vasculaires cérébraux (AVC), comparaison deux populations ayant eu soit un nouveau traitement, soit le traitement standard, avec un HR de 0,85 (IC95%: 0,65–1,10) → l’IC contient 1 → pas de supériorité démontrée du nouveau traitement.
L’analyse principale est généralement réalisée en intention de traiter, c’est-à-dire que les participants sont analysés selon le groupe auquel ils ont été initialement randomisés, indépendamment du traitement effectivement reçu, ce qui permet de préserver les bénéfices de la randomisation et d’obtenir une estimation pragmatique de l’effet du traitement. Lorsque des analyses intermédiaires sont prévues, elles doivent être spécifiées à l’avance et intégrer des méthodes d’ajustement du risque alpha afin de limiter l’inflation du risque d’erreur de type I (voir notre article sur la p-valeur). Une analyse intermédiaire peut être nécessaire pour évaluer une efficacité majeure, un défaut évident d’efficacité (futilité), un problème de tolérance ou encore une révision de la taille de l’échantillon. Dans ce cas, la poursuite de l’essai peut poser des questions éthiques et conduire à un arrêt anticipé (arrêt précoce).
- Le risque α (alpha) correspond au seuil de significativité statistique, généralement fixé à 0,05 dans les essais cliniques. Cela signifie que les chercheurs acceptent un risque maximal de 5% de conclure à tort qu’il existe une différence (erreur de type I). Dire qu’il n’y a pas de différence significative au risque αsignifie donc que la p-valeur est supérieure au seuil choisi (souvent p > 0,05).
- Attention, cela ne signifie pas nécessairement que les deux traitements sont équivalents, mais indique seulement que l’étude n’a pas mis en évidence de différence, ce qui peut aussi être dû à une puissance statistique insuffisante, une taille d’échantillon trop faible ou un effet réel mais trop modeste pour être détecté.
Etude de non-infériorité
Le design de non-infériorité répond à une problématique différente. Il ne s’agit plus de démontrer que le nouveau traitement est meilleur, mais qu’il n’est pas cliniquement inférieur au traitement de référence au-delà d’une perte d’efficacité jugée cliniquement acceptable. Ce type d’étude est particulièrement pertinent lorsque l’utilisation d’un placebo n’est pas éthiquement acceptable, notamment lorsqu’il existe un traitement de référence efficace validé. Il est également choisi lorsque le nouveau traitement présente d’autres avantages, tels qu’une meilleure tolérance, une plus grande facilité d’administration ou un coût réduit. Par conséquent, le choix de ce design est guidé par la balance entre l’efficacité et le gain pour le patient sur d’autres aspects du nouveau traitement testé.
La définition de la marge de non-infériorité, notée M, constitue l’élément central de ce design. Cette marge doit être fixée avant le début de l’étude, reposer sur des données historiques solides concernant l’efficacité du traitement de référence par rapport au placebo et être statistiquement valide. Elle doit représenter une proportion cliniquement pertinente de l’effet démontré antérieurement, ni trop faible, ni trop élevée. Sa détermination implique un dialogue étroit entre cliniciens et biostatisticiens (document de la FDA «Non-Inferiority Clinical Trials to Establish Effectiveness – Guidance for Industry»).
D’un point de vue statistique, les hypothèses qui sont à tester dans l’étude sont les suivantes :
- Nulle: la différence entre le traitement de contrôle et le traitement testé est supérieure ou égale à la marge de non-infériorité M;
- Alternative : la différence est strictement inférieure à la marge M.
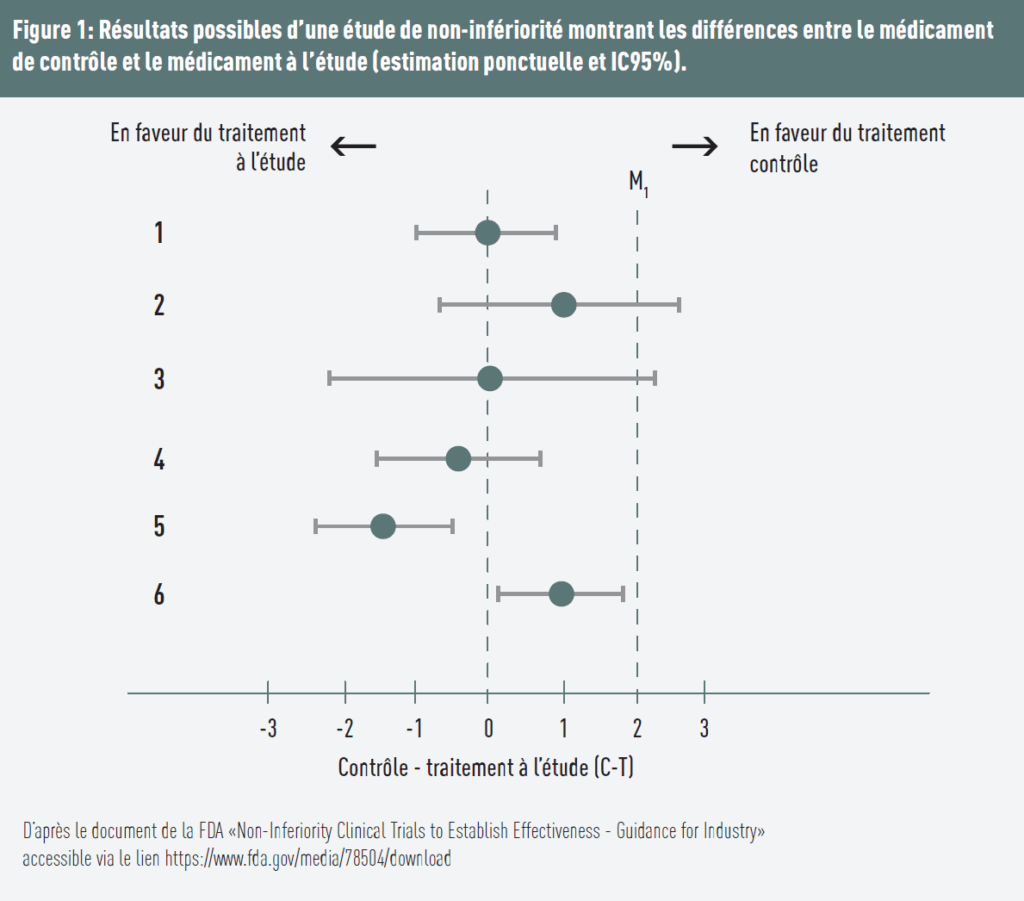
Plusieurs scénarios sont présentés sur la figure 1:
- la différence est égale à 0, supposant que l’effet des deux traitements est identique. La borne supérieure de l’intervalle de confiance (IC) à 95% est en dessous du seuil déterminé, alors la non-infériorité est démontrée;
- le traitement de contrôle est plus favorable, et la borne supérieure de l’intervalle de confiance est supérieure au seuil → la non-infériorité n’est pas démontrée;
- la différence est égale à 0, mais comme la borne supérieure de l’intervalle de confiance est supérieure à la marge → la non-infériorité n’est pas démontrée;
- le traitement testé est favorisé (le point d’estimation est inférieur à 0) et la borne supérieure de l’IC inférieure à la marge → la non-infériorité est démontrée;
- le traitement testé est favorisé, et comme la borne supérieure est inférieure à la marge et à 0, la non-infériorité et la supériorité sont démontrées;
- le traitement de référence apparaît plus efficace (estimation positive), mais l’intervalle de confiance reste entièrement inférieur à la marge Δ: la non-infériorité est donc démontrée. Cependant, la borne inférieure de l’IC étant supérieure à 0, les résultats suggèrent que le traitement testé est statistiquement inférieur au traitement de contrôle.
Les recommandations de l’agence européenne du médicament (EMA) et la US Food and Drug Administration (FDA) soulignent l’importance de conduire l’analyse en intention de traiter (ITT). Dans les essais de supériorité, l’analyse en intention de traiter (ITT) tend à atténuer les différences entre les groupes et constitue donc une approche conservatrice. À l’inverse, dans les essais de non-infériorité, cet adoucissement peut artificiellement favoriser la conclusion de non-infériorité. C’est pourquoi les recommandations méthodologiques préconisent généralement une analyse per-protocol (PP) et une analyse ITT.
Le calcul de la taille d’échantillon dans ce type d’étude dépend fortement de la marge retenue. Plus celle-ci est petite, plus le nombre de participants nécessaires sera important afin de garantir une puissance statistique suffisante. La taille de l’échantillon est aussi dépendante d’autres facteurs comme la taille de l’effet, le nombre de centres inclus dans l’étude… Mais ceci est une autre histoire !
Autrement dit: M correspond à la perte maximale d’efficacité du nouveau traitement par rapport au traitement de référence, compatible avec la préservation d’une fraction cliniquement pertinente de l’effet de ce dernier. Elle constitue donc la limite statistique en-deçà de laquelle le nouveau traitement serait jugé cliniquement inacceptable. Tant que l’effet estimé ne franchit pas cette limite, la non-infériorité (ou l’équivalence) peut être conclue.
CONSTRUCTION EN PRATIQUE:
- une première étape repose sur une analyse bibliographique des données d’efficacité du traitement de référence, généralement issues d’essais versus placebo;
- les estimations d’effet et leurs intervalles de confiance sont ensuite combinés;
- la marge est définie soit à partir de l’effet groupé, soit à partir de la borne de l’intervalle de confiance la plus proche de l’effet nul.
D’autres méthodes existent, mais elles sont plus complexes.
Etude d’équivalence
L’étude d’équivalence vise à démontrer que l’effet du nouveau traitement est suffisamment proche de celui du traitement de référence pour être considéré comme équivalent sur le plan clinique. Cela se traduit de manière statistique par une différence entre les deux traitements, comprise dans un intervalle prédéfini allant de –M à +M (voir la définition de la marge M décrite dans l’étude de non-infériorité). L’objectif est donc d’établir que les deux traitements ne diffèrent statistiquement pas, ni dans un sens ni dans l’autre.
Dans ce cadre, l’hypothèse nulle consiste à observer une différence située en dehors de l’intervalle d’équivalence, tandis que l’hypothèse alternative correspond à une différence incluse à l’intérieur de cet intervalle. L’équivalence est démontrée lorsque l’ensemble de l’intervalle de confiance à 95% se situe dans les limites fixées a priori. Le test est mené de manière bilatérale, ce qui le distingue d’une analyse de non-infériorité.
Ce design est fréquemment utilisé lors de l’évaluation de nouvelles formulations, ou de modifications de voie d’administration. Il convient de souligner qu’une étude de supériorité n’ayant pas mis en évidence de différence statistiquement significative ne permet en aucun cas de conclure à une équivalence. L’absence de preuve de différence ne constitue pas une preuve d’absence de différence, en particulier lorsque l’étude n’a pas été dimensionnée pour tester cette hypothèse spécifique.
L’interprétation correcte des essais comparatifs nécessite une collaboration étroite entre cliniciens et biostatisticiens dès la conception de l’étude.
D’AUTRES DESIGNS EXISTENT
Les schémas d’essais cliniques ne se limitent pas aux modèles classiques présentés dans cet article. Dans certaines situations, des approches spécifiques sont utilisées pour mieux répondre à la question de recherche. Par exemple, dans un essai «cross-over», chaque patient reçoit successivement le traitement et le placebo: il sert ainsi de propre témoin, ce qui permet de réduire la variabilité entre individus. Dans un essai factoriel, plusieurs interventions sont évaluées simultanément, seules ou en combinaison, afin d’étudier à la fois leurs effets propres et leurs éventuelles interactions.
Le développement d’un nouveau médicament ou vaccin reste un processus long, généralement de l’ordre de 15 à 20 ans dans un cadre standard. Pour accélérer ce processus, des designs plus flexibles ont été développés. Certains consistent à regrouper ou à faire se chevaucher plusieurs phases d’essais (par exemple phases I–II ou II–III), afin de gagner du temps sans compromettre l’évaluation de la sécurité et de l’efficacité. Cette approche a été largement mise en oeuvre lors du développement des vaccins à ARNm contre la COVID-19, où les phases précoces ont été imbriquées et la phase III initiée très rapidement sur la base de données intermédiaires. D’autres designs, dits «adaptatifs», permettent quant à eux d’ajuster certains éléments de l’étude en cours de route sur la base d’analyses intermédiaires (par exemple modifier les doses, réallouer les patients ou arrêter précocement un bras inefficace).
La pandémie de COVID-19 a également contribué à populariser les essais «de plateforme». Ces essais reposent sur une infrastructure commune dans laquelle plusieurs traitements peuvent être évalués en parallèle, avec la possibilité d’en ajouter ou d’en retirer au fil du temps. Ce type d’organisation améliore considérablement l’efficacité et la réactivité de la recherche clinique.
Enfin, le développement de la médecine personnalisée a conduit à l’émergence de designs innovants. Les essais «basket» évaluent un même traitement ciblé dans différentes maladies partageant une même anomalie moléculaire. À l’inverse, les essais «umbrella» testent plusieurs traitements ciblés au sein d’une seule maladie, en fonction des caractéristiques moléculaires propres à des sous-groupes de patients. Ces approches permettent d’adapter plus finement les traitements au profil biologique des patients.
Conclusions
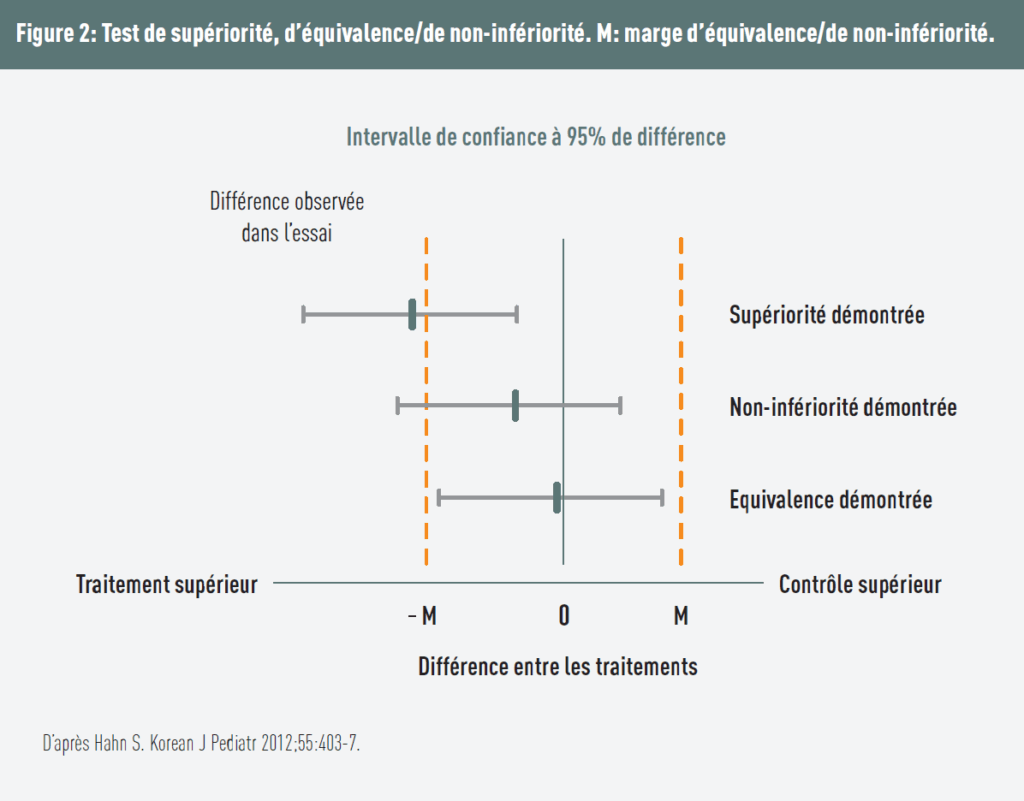
La figure 2 permet de distinguer les différents schémas.
La marge M ne doit pas être confondue avec la significativité statistique: il s’agit de deux concepts totalement différents. La marge est définie à partir des données de la littérature, de l’expérience acquise et des discussions avec les cliniciens; elle est difficile à déterminer. Les conclusions des essais de non-infériorité et d’équivalence se basent exclusivement sur cette marge. Une marge trop large peut conduire à accepter une perte d’efficacité cliniquement inacceptable, tandis qu’une marge trop stricte rendra la démonstration statistique particulièrement exigeante.
Le choix du risque alpha détermine les bornes de l’intervalle de confiance (le risque alpha peut être 5% et donc l’intervalle de confiance est à 95%). Dans une étude de non-infériorité on pourra choisir un alpha à 1% (et une puissance à 90% au lieu de 80%) afin d’adopter une approche plus conservatrice (IC à 99%). La non-infériorité devient plus difficile à démontrer mais le risque de faux positif est diminué i.e. le nouveau traitement a moins de chance de ne pas être non-inférieur au traitement de référence.
Le choix du design doit impérativement être déterminé avant le début de l’étude, car il conditionne la formulation des hypothèses, le calcul de la taille d’échantillon, les modalités d’analyse et l’interprétation des résultats. La question centrale reste avant tout clinique:
- mon nouveau traitement est-il meilleur que le traitement existant ? → étude de supériorité
- mon nouveau traitement est-il presque aussi efficace, tout en offrant d’autres avantages? → étude de non-infériorité
- mon nouveau traitement est-il cliniquement équivalent au traitement existant ? → étude d’équivalence
Compte tenu de la complexité méthodologique et réglementaire de ces essais comparatifs, l’implication en amont de l’étude d’un biostatisticien est essentielle afin d’assurer la validité scientifique et la robustesse des conclusions.
Take home messages
- 1. Le choix du design d’essai est d’abord une décision clinique. Il dépend de la question posée: démontrer une supériorité, préserver l’efficacité existante ou montrer une équivalence.
- 2. L’absence de différence statistiquement significative ne signifie pas que deux traitements sont équivalents. Une étude de supériorité négative ne permet pas de conclure à une équivalence ou à une non-infériorité.
- 3. Dans les essais de non-infériorité et d’équivalence, la définition de la marge Δ est une étape critique. Une marge trop large peut conduire à accepter une perte d’efficacité cliniquement inacceptable.
- 4. Les conclusions des essais de non-infériorité et d’équivalence reposent essentiellement sur l’intervalle de confiance et la marge de non-infériorité/ d’équivalence.
- 5. Dans les essais de non-infériorité, les analyses per-protocol et en intention de traiter doivent généralement être concordantes pour renforcer la robustesse des conclusions.
- 6. L’interprétation correcte des essais comparatifs nécessite une collaboration étroite entre cliniciens et biostatisticiens dès la conception de l’étude.
The epidemiostatistical series published in MEDINLUX.

Actualités associées