News
Keine Scheu vor Überlegenheits-, Nicht-Unterlegenheits- und Äquivalenzstudien!
Die Wissenschaft der Daten verstehen
Dieser Artikel wurde in MedinLux veröffentlicht und ist Teil einer gemeinsamen Anstrengung, statistische und epidemiologische Konzepte den Angehörigen der Gesundheitsberufe in Luxemburg zugänglich zu machen.
In einem unserer vorherigen Artikel haben wir die grundlegenden Prinzipien des Designs klinischer Studien beschrieben. Hier beschäftigen wir uns mit drei Studientypen, die hauptsächlich in klinischen Studien der Phase III verwendet werden: Überlegenheits-, Nichtunterlegenheits- und Äquivalenzstudien.
Phase-III-Studien sind vergleichende Studien, in der Regel randomisiert, an denen mehrere hundert bis mehrere tausend Teilnehmende beteiligt sind. Ihr Ziel ist es, die Wirksamkeit und Sicherheit einer neuen Behandlung oder eines neuen Medizinprodukts im großen Maßstab im Vergleich zu einer Referenzbehandlung oder einem Placebo zu bewerten. Wenn die Ergebnisse als positiv angesehen werden, bilden sie die Grundlage des Zulassungsantrags bei den zuständigen Behörden.
Die Wahl des Studiendesigns bestimmt das gesamte Protokoll: statistische Hypothesen, Berechnung der Stichprobengröße, Analysemethoden und Interpretation der Ergebnisse.
„Outcomes“ und „Endpoints“: Definitionen und Herausforderungen
Zunächst ist die präzise Definition der Bewertungskriterien essenziell. Diese Elemente werden im Protokoll beschrieben und im statistischen Analyseplan detailliert dargestellt. Obwohl die Begriffe „Outcome“ und „Endpoint“ häufig synonym verwendet werden, beziehen sie sich auf unterschiedliche Konzepte:
- „Outcome“: jede Messgröße, die mit dem Gesundheitszustand eines Teilnehmenden zusammenhängt (Depressionsscore, Tumorgröße, Blutdruck, Auftreten eines kardiovaskulären Ereignisses usw.).
- „Endpoint“ (Bewertungskriterium): operationelle und zeitliche Definition des „Outcomes“, die zur Beantwortung des primären Studienziels dient (zum Beispiel: Senkung des systolischen Blutdrucks zwischen Studienbeginn und der zwölften Woche).
Ein relevanter „Endpoint“ muss einen klaren klinischen Nutzen aufweisen und ausreichend präzise definiert sein, um die Reproduzierbarkeit der Messung zu gewährleisten. Es muss mit der Berechnung der Stichprobengröße übereinstimmen und, wenn möglich, auf validierten Instrumenten beruhen. Die a-priori-Definition des primären Bewertungskriteriums ist essenziell, um jede nachträgliche Interpretation der Ergebnisse zu vermeiden, die deren Auslegung verfälschen würde. Die Art des „Endpoints“ (kontinuierlich, binär, Zeit bis zum Ereignis) beeinflusst ebenfalls die Wahl der statistischen Methoden und des Studiendesigns.
Überlegenheitsstudie
Die Überlegenheitsstudie stellt das klassischste Design in der klinischen Forschung dar. Ihr Ziel ist es zu zeigen, dass die neue Behandlung dem Vergleichspräparat statistisch überlegen ist, unabhängig davon, ob es sich um ein Placebo oder eine aktive Behandlung handelt.
In diesem Rahmen werden die in der Studie geprüften statistischen Hypothesen wie folgt definiert:
- Nullhypothese: Es besteht kein statistisch signifikanter Unterschied, gemessen anhand des „Endpoints“, zwischen der neuen Behandlung und der Kontrollbehandlung beim gewählten Alpha-Risiko (siehe unseren Artikel zum p-Wert und den Infokasten);
- Alternativhypothese: Der Unterschied zwischen den beiden Behandlungen ist statistisch signifikant.
Die Analyse erfolgt zweiseitig mit 95%-Konfidenzintervallen (95%-KI). Die Überlegenheit gilt als nachgewiesen, wenn dieses Konfidenzintervall den Wert, der dem Fehlen eines Effekts entspricht, nicht einschließt (0 für eine absolute Differenz, 1 für ein Verhältnis wie relatives Risiko, Odds Ratio oder Hazard Ratio). Einige fiktive Beispiele:
- eine mittlere Differenz des systolischen Blutdrucks von 6 mmHg (95%-KI: -10; -2) nach Einnahme einer Behandlung → das KI enthält 0 nicht → statistisch signifikanter Unterschied bei einem Risiko von 5 %;
- ein Überlebensvergleich nach einer Krebsdiagnose zwischen einer neuen Behandlung und der Standardbehandlung mit einer Hazard Ratio (HR) von 0,78 (95%-KI: 0,65–0,92) → das KI enthält 1 nicht → signifikante Risikoreduktion unter der Behandlung;
- ein Vergleich des Auftretens von Schlaganfällen zwischen einer neuen Behandlung und der Standardbehandlung mit einer HR von 0,85 (95%-KI: 0,65–1,10) → das KI enthält 1 → keine nachgewiesene Überlegenheit der neuen Behandlung.
Die Hauptanalyse wird im Allgemeinen nach dem „Intention-to-treat“-Prinzip durchgeführt, das heißt, die Teilnehmenden werden entsprechend der Gruppe analysiert, der sie ursprünglich randomisiert wurden, unabhängig von der tatsächlich erhaltenen Behandlung. Dadurch bleiben die Vorteile der Randomisierung erhalten und es wird eine pragmatische Schätzung des Behandlungseffekts ermöglicht. Wenn Zwischenanalysen vorgesehen sind, müssen diese im Voraus festgelegt werden und Methoden zur Anpassung des Alpha-Risikos enthalten, um eine Inflation des Risikos eines Fehlers erster Art zu begrenzen (siehe unseren Artikel zum p-Wert). Eine Zwischenanalyse kann notwendig sein, um eine deutliche Wirksamkeit, eine offensichtliche mangelnde Wirksamkeit („Futility“), ein Verträglichkeitsproblem oder eine Anpassung der Stichprobengröße zu bewerten. In diesem Fall kann die Fortführung der Studie ethische Fragen aufwerfen, was zu einem vorzeitigen Studienabbruch führen kann.
- Das Risiko α (Alpha) entspricht der Schwelle für statistische Signifikanz, das in klinischen Studien üblicherweise auf 0,05 festgelegt wird. Das bedeutet, dass die Forschenden ein maximales Risiko von 5 % akzeptieren, fälschlicherweise eine Differenz zu behaupten (Fehler 1. Art). Zu sagen, dass kein signifikanter Unterschied beim Risiko α besteht, bedeutet somit, dass der p-Wert über dem gewählten Schwellenwert liegt (oft p > 0,05).
- Achtung: Dies bedeutet nicht zwangsläufig, dass die beiden Behandlungen äquivalent sind, sondern lediglich, dass die Studie keinen Unterschied nachweisen konnte. Dies kann auch auf eine unzureichende Teststärke („Power“), eine zu kleine Stichprobengröße oder einen zu kleinen realen Effekt für einen erfolgreichen Nachweis zurückzuführen sein.
Nichtunterlegenheitsstudie
Das Design der Nicht-Unterlegenheitsstudie beantwortet eine andere Fragestellung. Es geht nicht mehr darum zu zeigen, dass die neue Behandlung besser ist, sondern dass sie dem Referenzstandard klinisch nicht unterlegen ist, abgesehen von einem als klinisch akzeptabel erachteten Wirkungsverlust. Dieses Studiendesign ist besonders relevant, wenn der Einsatz eines Placebos ethisch nicht vertretbar ist, insbesondere wenn eine wirksame Standardbehandlung bereits existiert. Es wird auch gewählt, wenn die neue Behandlung andere Vorteile bietet, wie eine bessere Verträglichkeit, eine einfachere Anwendung oder geringere Kosten. Die Wahl dieses Designs ergibt sich somit aus der Abwägung zwischen Wirksamkeit und zusätzlichen patientenrelevanten Vorteilen.
Die Definition der Nichtunterlegenheitsgrenze, bezeichnet als M, ist das zentrale Element dieses Designs. Diese Marge muss vor Studienbeginn festgelegt werden, auf soliden historischen Daten zur Wirksamkeit der Referenzbehandlung gegenüber Placebo beruhen und statistisch valide sein. Sie muss einen klinisch relevanten Anteil des zuvor nachgewiesenen Effekts darstellen, weder zu klein noch zu groß. Ihre Festlegung erfordert eine enge Zusammenarbeit zwischen Klinikern und Biostatistikern (FDA-Dokument: Non-Inferiority Clinical Trials to Establish Effectiveness – Guidance for Industry).
Aus statistischer Sicht werden folgende Hypothesen geprüft:
- Nullhypothese: Die Differenz zwischen Kontroll- und Testbehandlung ist größer oder gleich der Nichtunterlegenheitsgrenze M;
Alternativhypothese: Die Differenz ist strikt kleiner als M.
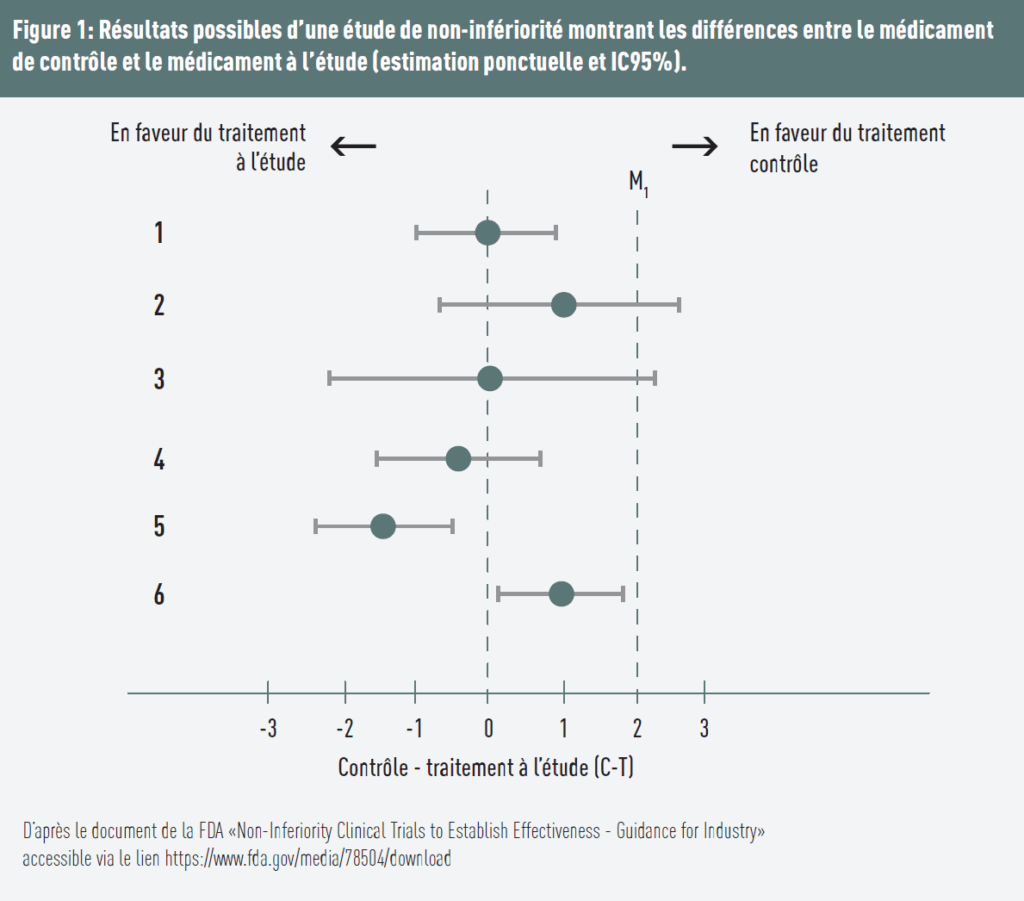
Mehrere Szenarien sind in Abbildung 1 dargestellt:
- Die Differenz ist gleich 0, was eine identische Wirkung beider Behandlungen impliziert. Die obere Grenze des 95%-Konfidenzintervalls liegt unterhalb der festgelegten Grenze → Nichtunterlegenheit ist gezeigt;
- Die Kontrollbehandlung ist überlegen, und die obere Grenze des Konfidenzintervalls liegt über der Grenze → Nichtunterlegenheit ist nicht gezeigt;
- Die Differenz ist gleich 0, aber die obere Grenze des Konfidenzintervalls liegt über der Grenze → Nichtunterlegenheit ist nicht gezeigt;
- Die Testbehandlung ist im Vorteil (Schätzpunkt < 0) und die obere Grenze des Konfidenzintervalls liegt unterhalb der Grenze → Nichtunterlegenheit ist gezeigt;
- Die Testbehandlung ist im Vorteil und die obere Grenze liegt sowohl unter der Grenze als auch unter 0 → Nichtunterlegenheit und Überlegenheit sind gezeigt;
- Die Referenzbehandlung erscheint wirksamer (Schätzpunkt > 0), aber das gesamte Konfidenzintervall liegt unterhalb der Grenze → Nichtunterlegenheit ist gezeigt. Da jedoch die untere Grenze des Konfidenzintervalls über 0 liegt, deuten die Ergebnisse zudem auf eine statistische Unterlegenheit der Testbehandlung hin.
Die Empfehlungen der Europäischen Arzneimittel-Agentur (EMA) und der US Food and Drug Administration (FDA) betonen die Bedeutung der „Intention-to-treat“-Analyse (ITT). In Überlegenheitsstudien ist die ITT-Analyse konservativ, da sie Unterschiede zwischen Gruppen eher abschwächt. In Nichtunterlegenheitsstudien kann diese Abschwächung jedoch fälschlich zur Schlussfolgerung der Nichtunterlegenheit führen. Daher empfehlen methodische Leitlinien in der Regel sowohl eine „Per-Protocol“-Analyse (PP) als auch eine ITT-Analyse. Die Berechnung der Stichprobengröße hängt in diesem Design stark von der gewählten Grenze M ab. Je kleiner diese ist, desto größer muss die Anzahl der Teilnehmenden sein, um eine ausreichende statistische Power zu gewährleisten. Die Stichprobengröße hängt außerdem von weiteren Faktoren ab, wie der Effektstärke oder der Anzahl der beteiligten Zentren… Doch das ist eine andere Geschichte.
Anders gesagt entspricht M der maximal akzeptablen Wirksamkeitsminderung der neuen Behandlung im Vergleich zur Referenzbehandlung, die noch mit der Erhaltung eines klinisch relevanten Anteils des Effekts dieser Referenzbehandlung vereinbar ist. Sie stellt somit die statistische Grenze dar, unterhalb derer die neue Behandlung als klinisch inakzeptabel gelten würde. Solange der geschätzte Effekt diese Grenze nicht überschreitet, kann Nichtunterlegenheit (oder Äquivalenz) angenommen werden.
DESIGN IN DER PRAXIS:
- ein erster Schritt besteht in einer systematischen Literaturrecherche zu den Wirksamkeitsdaten der Referenzbehandlung, in der Regel aus Placebo-kontrollierten Studien;
- die Schätzungen des Effekts und ihre Konfidenzintervalle werden anschließend zusammengeführt;
die Marge wird entweder auf Basis des gepoolten Effekts oder auf Grundlage der Konfidenzintervallgrenze festgelegt, die dem Null-Effekt am nächsten liegt.
Weitere Methoden existieren, sind jedoch komplexer.
Äquivalenzstudie
Die Äquivalenzstudie zielt darauf ab zu zeigen, dass die Wirkung der neuen Behandlung der Referenzbehandlung ausreichend nahekommt, um klinisch als äquivalent angesehen zu werden. Statistisch äußert sich dies darin, dass die Differenz zwischen beiden Behandlungen innerhalb eines vordefinierten Intervalls von –M bis +M liegt (siehe Definition der Grenze M in der Nicht-Unterlegenheitsstudie). Ziel ist es somit, zu zeigen, dass sich die beiden Behandlungen weder in die eine noch in die andere Richtung statistisch unterscheiden.
In diesem Rahmen besteht die Nullhypothese darin, eine Differenz außerhalb des Äquivalenzintervalls zu beobachten, während die Alternativhypothese einer Differenz innerhalb dieses Intervalls entspricht. Äquivalenz ist gezeigt, wenn das gesamte 95%-Konfidenzintervall innerhalb der a priori festgelegten Grenzen liegt. Der Test wird zweiseitig durchgeführt, was ihn von der Nicht-Unterlegenheitsanalyse unterscheidet. Dieses Design wird häufig bei der Bewertung neuer Formulierungen oder Änderungen der Applikationsform verwendet. Es ist wichtig zu betonen, dass eine Überlegenheitsstudie, die keinen statistisch signifikanten Unterschied zeigt, keinesfalls den Schluss auf Äquivalenz erlaubt. Das Fehlen von Nachweisen für einen Unterschied ist kein Beweis dafür, dass kein Unterschied besteht, insbesondere wenn die Studie nicht darauf ausgelegt war, diese spezifische Hypothese zu überprüfen.
Die korrekte Interpretation vergleichender Studien erfordert eine enge Zusammenarbeit zwischen Klinikern und Biostatistikern bereits in der Planungsphase der Studie.
ANDERE STUDIENDESIGNS EXISTIEREN
Die Designs klinischer Studien beschränken sich nicht auf die in diesem Artikel dargestellten klassischen Modelle. In bestimmten Situationen werden spezifische Ansätze verwendet, um die Forschungsfrage besser zu beantworten. So erhält in einer „Cross-over“-Studie jeder Patient nacheinander die Behandlung und das Placebo: Er dient somit als seine eigene Kontrollgruppe, wodurch die Variabilität zwischen den einzelnen Personen verringert wird. In einer faktoriellen Studie werden mehrere Interventionen gleichzeitig – einzeln oder in Kombination – untersucht, um sowohl ihre jeweiligen Wirkungen als auch mögliche Wechselwirkungen zu untersuchen.
Die Entwicklung eines neuen Medikaments oder Impfstoffs ist nach wie vor ein langwieriger Prozess, der unter normalen Umständen etwa 15 bis 20 Jahre dauert. Um diesen Prozess zu beschleunigen, wurden flexiblere Designs entwickelt. Einige bestehen darin, mehrere Studienphasen zusammenzuführen oder zu überlappen (zum Beispiel Phase I–II oder II–III), um Zeit zu gewinnen, ohne die Bewertung von Sicherheit und Wirksamkeit zu beeinträchtigen. Dieser Ansatz wurde während der Entwicklung der mRNA-Impfstoffe gegen COVID-19 breit eingesetzt, wobei frühe Phasen überlappt wurden und Phase III sehr schnell auf Grundlage intermediärer Daten gestartet wurde. Andere sogenannte „adaptive Designs“ ermöglichen es, bestimmte Studienelemente während der laufenden Studie auf Basis von Zwischenanalysen anzupassen (zum Beispiel Dosierungen zu ändern, Patienten neu zuzuweisen oder einen ineffektiven Arm vorzeitig zu beenden).
Die COVID-19-Pandemie hat auch zur Popularisierung sogenannter „Plattform“-Studien beigetragen. Diese basieren auf einer gemeinsamen Infrastruktur, in der mehrere Behandlungen parallel bewertet werden können, mit der Möglichkeit, neue Behandlungen hinzuzufügen oder bestehende im Verlauf zu entfernen. Dieses Design verbessert die Effizienz und Reaktionsfähigkeit der klinischen Forschung erheblich.
Schließlich hat auch die Entwicklung der personalisierten Medizin zur Entstehung innovativer Designs geführt. „Basket“-Studien bewerten eine zielgerichtete Therapie bei verschiedenen Erkrankungen, die eine gemeinsame molekulare Anomalie aufweisen. Im Gegensatz dazu werden bei „Umbrella“-Studien mehrere zielgerichtete Therapien bei einer einzigen Erkrankung getestet, je nach den molekularen Merkmalen bestimmter Patientengruppen. Diese Ansätze ermöglichen eine präzisere Anpassung der Behandlung an das biologische Profil der Patienten.
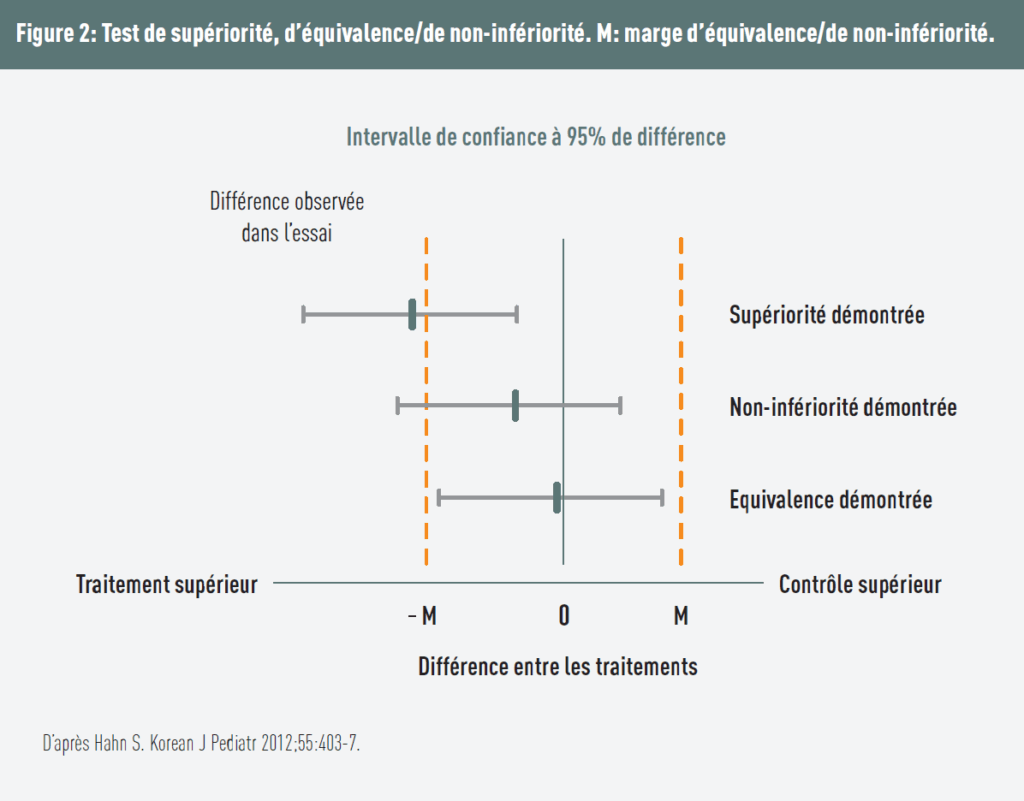
Schlussfolgerungen
Abbildung 2 ermöglicht die Unterscheidung der verschiedenen Studiendesigns.
Die Grenze M darf nicht mit statistischer Signifikanz verwechselt werden: Es handelt sich um zwei völlig unterschiedliche Konzepte. Die Grenze wird auf Basis von Literaturdaten, klinischer Erfahrung und Diskussionen mit den Klinikern definiert; sie ist schwer festzulegen. Die Schlussfolgerungen von Nicht-Unterlegenheits- und Äquivalenzstudien basieren ausschließlich auf dieser Grenze. Ist sie zu großzügig gewählt, kann das dazu führen, dass ein klinisch inakzeptabler Wirksamkeitsverlust akzeptiert wird, während eine zu strenge Marge den statistischen Nachweis besonders anspruchsvoll macht.
Die Wahl des Alpha-Risikos bestimmt die Grenzen des Konfidenzintervalls (ein Alpha-Risiko von 5 % entspricht einem 95%-Konfidenzintervall). In einer Nicht-Unterlegenheitsstudie kann ein Alpha von 1 % (und eine Power von 90 % statt 80 %) gewählt werden, um einen konservativeren Ansatz zu verfolgen (99%-Konfidenzintervall). Dadurch wird die Nicht-Unterlegenheit schwieriger nachzuweisen, aber das Risiko eines falsch-positiven Ergebnisses sinkt, d. h. die Wahrscheinlichkeit, dass eine neue Behandlung fälschlicherweise als nicht unterlegen eingestuft wird, ist geringer.
Die Wahl des Designs muss zwingend vor Beginn der Studie festgelegt werden, da sie die Formulierung der Hypothesen, die Berechnung der Stichprobengröße, die Analysemethoden und die Interpretation der Ergebnisse bestimmt. Die zentrale Fragestellung bleibt vor allem klinisch:
- Ist meine neue Behandlung besser als die bestehende Behandlung? → Überlegenheitsstudie
- Ist meine neue Behandlung nahezu gleich wirksam, bietet aber andere Vorteile? → Nicht-Unterlegenheitsstudie
- Ist meine neue Behandlung klinisch äquivalent zur bestehenden Behandlung? → Äquivalenzstudie
Angesichts der methodischen und regulatorischen Komplexität dieser vergleichenden Studien ist die frühzeitige Einbindung eines Biostatistikers entscheidend, um die wissenschaftliche Validität und die Robustheit der Schlussfolgerungen sicherzustellen.
Take-home messages
- Die Wahl des Studiendesigns ist in erster Linie eine klinische Entscheidung. Sie hängt von der Fragestellung ab: Überlegenheit nachweisen, bestehende Wirksamkeit erhalten oder Äquivalenz zeigen.
- Das Fehlen eines statistisch signifikanten Unterschieds bedeutet nicht, dass zwei Behandlungen äquivalent sind. Eine negative Überlegenheitsstudie erlaubt keinen Schluss auf Äquivalenz oder Nicht-Unterlegenheit.
- In Nicht-Unterlegenheits- und Äquivalenzstudien ist die Definition der Grenze Δ[MB1] ein kritischer Schritt. Eine zu große Grenze kann dazu führen, dass ein klinisch inakzeptabler Wirksamkeitsverlust akzeptiert wird.
- Die Schlussfolgerungen von Nicht-Unterlegenheits- und Äquivalenzstudien beruhen im Wesentlichen auf dem Konfidenzintervall und der Nicht-Unterlegenheits- bzw. Äquivalenzgrenze.
- In Nicht-Unterlegenheitsstudien sollten „Per-Protocol“- und „Intention-to-treat“-Analysen in der Regel übereinstimmen, um die Robustheit der Schlussfolgerungen zu stärken.
- Die korrekte Interpretation vergleichender Studien erfordert eine enge Zusammenarbeit zwischen Klinikern und Biostatistikern bereits in der Planungsphase der Studie.
Die in MEDINLUX veröffentlichten epidemiostatistischen Reihen.

Ähnliche News